#+TITLE: COVID-19 PubSeq - Arvados
#+AUTHOR: Pjotr Prins
# C-c C-e h h publish
# C-c ! insert date (use . for active agenda, C-u C-c ! for date, C-u C-c . for time)
# C-c C-t task rotate
# RSS_IMAGE_URL: http://xxxx.xxxx.free.fr/rss_icon.png
#+HTML_LINK_HOME: http://covid19.genenetwork.org
#+HTML_HEAD:
* Table of Contents :TOC:noexport:
- [[#the-arvados-web-server][The Arvados Web Server]]
- [[#the-arvados-file-interface][The Arvados file interface]]
- [[#the-pubseq-arvados-shell][The PubSeq Arvados shell]]
- [[#wiring-up-cwl][Wiring up CWL]]
- [[#using-the-arvados-api][Using the Arvados API]]
- [[#troubleshooting][Troubleshooting]]
* The Arvados Web Server
We are using Arvados to run common workflow language (CWL) pipelines.
The most recent output is on display on a [[https://workbench.lugli.arvadosapi.com/collections/lugli-4zz18-z513nlpqm03hpca][web page]] (with time stamp)
and a full output list is generated [[https://collections.lugli.arvadosapi.com/c=lugli-4zz18-z513nlpqm03hpca/][here]].
Arvados has a web front which allows navigation through input and output data,
workflows and the output of analysis pipelines (here CWL workflows).
@@html: @@
* The Arvados file interface
Arvados has the web server, but it also has a REST API and associated
command line tools. We are already using the [[https://github.com/arvados/bh20-seq-resource/blob/master/bh20sequploader/main.py#L27][API]] to upload data. If
you follow the pip or [[../INSTALL.md]] GNU Guix instructions for
installing Arvados API you'll find the following command line tools
(also documented [[https://doc.arvados.org/v2.0/sdk/cli/subcommands.html][here]]):
| Command | Description |
|---------+-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| arv-ls | list files in Arvados |
| arv-put | upload a file to Arvados |
| arv-get | get a textual representation of Arvados objects from the command line. The output can be limited to a subset of the object’s fields. This command can be used with only the knowledge of an object’s UUID |
Now, this is a public instance so we can use the tokens from
the [[https://github.com/arvados/bh20-seq-resource/blob/master/bh20sequploader/main.py#L16][uploader]].
#+BEGIN_SRC sh
export ARVADOS_API_HOST='lugli.arvadosapi.com'
export ARVADOS_API_TOKEN='2fbebpmbo3rw3x05ueu2i6nx70zhrsb1p22ycu3ry34m4x4462'
arv-ls lugli-4zz18-z513nlpqm03hpca
#+END_SRC
will list all files (the UUID we got from the Arvados results page). To
get the UUID of the files
#+BEGIN_SRC sh
curl https://lugli.arvadosapi.com/arvados/v1/config | jq .Users.AnonymousUserToken
env ARVADOS_API_TOKEN=5o42qdxpxp5cj15jqjf7vnxx5xduhm4ret703suuoa3ivfglfh \
arv-get lugli-4zz18-z513nlpqm03hpca
#+END_SRC
and fetch one listed JSON file ~chunk001_bin4000.schematic.json~ with
its listed UUID:
: arv-get 2be6af7b4741f2a5c5f8ff2bc6152d73+1955623+Ab9ad65d7fe958a053b3a57d545839de18290843a@5ed7f3c5
* The PubSeq Arvados shell
When you login to Arvados (you can request permission from us) it is
possible to upload an ssh key in your profile and get an shell prompt
with
: ssh pjotrpbl@shell.lugli.arvadosapi.com
: Linux ip-10-255-0-202 4.19.0-9-cloud-amd64 #1 SMP Debian 4.19.118-2+deb10u1 (2020-06-07) x86_64
It is a small Debian VM hosted on AWS somewhere. The PubSeq material
is mounted on ~/data/pubseq~. The log is in ~nohup.out~. Update/edit
the code (bh20-seq-resource git checkout) and restart the service (the
run script). The log says
: you should have permission to read the log (nohup.out) update / edit the code (bh20-seq-resource git checkout) and restart the service (the run script)
which means it will trigger the run on upload. The service is running as a
Python virtualenv:
: /data/pubseq/bh20-seq-resource/venv3/bin/python3 /data/pubseq/bh20-seq-resource/venv3/bin/bh20-seq-analyzer --no-start-analysis
and is restarted by a ~run~ script:
: /data/pubseq/run [options]
The run script kills the old process, sets up the API tokens, pulls
the git repo and starts a new run calling into
/data/pubseq/bh20-seq-resource/venv3/bin/bh20-seq-analyzer which is
essentially [[https://github.com/arvados/bh20-seq-resource/blob/2baa88b766ec540bd34b96599014dd16e393af39/bh20seqanalyzer/main.py#L354][monitoring]] for uploads.
On ~run --help~
#+begin_example
optional arguments:
-h, --help show this help message and exit
--uploader-project UPLOADER_PROJECT
--pangenome-analysis-project PANGENOME_ANALYSIS_PROJECT
--fastq-project FASTQ_PROJECT
--validated-project VALIDATED_PROJECT
--workflow-def-project WORKFLOW_DEF_PROJECT
--pangenome-workflow-uuid PANGENOME_WORKFLOW_UUID
--fastq-workflow-uuid FASTQ_WORKFLOW_UUID
--exclude-list EXCLUDE_LIST
--latest-result-collection LATEST_RESULT_COLLECTION
--kickoff
--no-start-analysis
--once
--print-status PRINT_STATUS
--revalidate
#+end_example
* Wiring up CWL
In above script ~bh20-seq-analyzer~ you can see that the [[https://www.commonwl.org/][Common
Workflow Language]] (CWL) gets [[https://github.com/arvados/bh20-seq-resource/blob/2baa88b766ec540bd34b96599014dd16e393af39/bh20seqanalyzer/main.py#L233][triggered]]; for example [[https://github.com/arvados/bh20-seq-resource/tree/master/workflows/fastq2fasta][fastq2fasta]] which
is part of the main repo. The actual script is in [[https://github.com/arvados/bh20-seq-resource/blob/master/workflows/fastq2fasta/fastq2fasta.cwl][fastq2fasta.cwl]] and
runs the following tools in sequence: bwa-mem, samtools-view,
samtools-sort, and bam2fasta.
It probably pays to familiarize yourself with CWL and its concepts. We
believe it has a lot going for it though CWL is some steps removed
from traditional shell scripts for running work flows. Main thing to
understand is that CWL is a separation of concerns, i.e.,
1. Data
2. Tools
3. Flow
and each of these are described separately. This contrasts largely
with shell scripts (though you can invoke shell scripts from CWL).
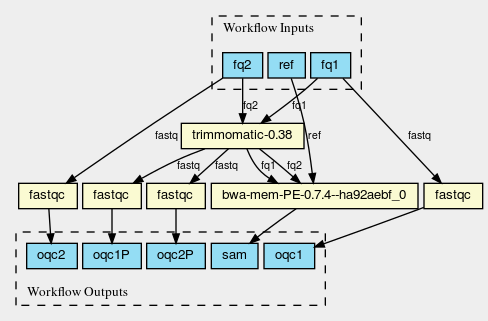
Also, CWL is written in JSON/YAML, which means everything can be parsed
as a tree and you can easily get visualisations such as
@@html: @@
For more see [[https://hpc.guix.info/blog/2019/01/creating-a-reproducible-workflow-with-cwl/][Creating a reproducible workflow with CWL]] by Pjotr Prins.
* Using the Arvados API
Arvados provides a rich API for accessing internals of the Cloud
infrastructure.
In above script ~bh20-seq-analyzer~ there are examples of querying the
[[https://doc.arvados.org/api/index.html][Arvados API]] using the [[https://pypi.org/project/arvados-python-client/][Python Arvados client and libraries]]. For example
get a list of [[https://github.com/arvados/bh20-seq-resource/blob/2baa88b766ec540bd34b96599014dd16e393af39/bh20seqanalyzer/main.py#L228][projects]] in Arvados. Main thing is to get the
~ARVADOS-API-HOST~ and ~ARVADOS-API-TOKEN~ right as is shown above.
* Troubleshooting
When workflows have errors we should check the logs in Arvados.
Go to the [[https://workbench.lugli.arvadosapi.com/projects/lugli-j7d0g-825x3r5vcs41dus][project]] page for 'COVID-19-BH20 Shared Project' -> 'Public
Sequence Resource'. Click on analysis runs
https://workbench.lugli.arvadosapi.com/projects/lugli-j7d0g-y4k4uswcqi3ku56
and 'Subprojects'. Click one of the runs and then on 'Processes' and you'll
see what parts failed.
 @@
* The Arvados file interface
Arvados has the web server, but it also has a REST API and associated
command line tools. We are already using the [[https://github.com/arvados/bh20-seq-resource/blob/master/bh20sequploader/main.py#L27][API]] to upload data. If
you follow the pip or [[../INSTALL.md]] GNU Guix instructions for
installing Arvados API you'll find the following command line tools
(also documented [[https://doc.arvados.org/v2.0/sdk/cli/subcommands.html][here]]):
| Command | Description |
|---------+-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| arv-ls | list files in Arvados |
| arv-put | upload a file to Arvados |
| arv-get | get a textual representation of Arvados objects from the command line. The output can be limited to a subset of the object’s fields. This command can be used with only the knowledge of an object’s UUID |
Now, this is a public instance so we can use the tokens from
the [[https://github.com/arvados/bh20-seq-resource/blob/master/bh20sequploader/main.py#L16][uploader]].
#+BEGIN_SRC sh
export ARVADOS_API_HOST='lugli.arvadosapi.com'
export ARVADOS_API_TOKEN='2fbebpmbo3rw3x05ueu2i6nx70zhrsb1p22ycu3ry34m4x4462'
arv-ls lugli-4zz18-z513nlpqm03hpca
#+END_SRC
will list all files (the UUID we got from the Arvados results page). To
get the UUID of the files
#+BEGIN_SRC sh
curl https://lugli.arvadosapi.com/arvados/v1/config | jq .Users.AnonymousUserToken
env ARVADOS_API_TOKEN=5o42qdxpxp5cj15jqjf7vnxx5xduhm4ret703suuoa3ivfglfh \
arv-get lugli-4zz18-z513nlpqm03hpca
#+END_SRC
and fetch one listed JSON file ~chunk001_bin4000.schematic.json~ with
its listed UUID:
: arv-get 2be6af7b4741f2a5c5f8ff2bc6152d73+1955623+Ab9ad65d7fe958a053b3a57d545839de18290843a@5ed7f3c5
* The PubSeq Arvados shell
When you login to Arvados (you can request permission from us) it is
possible to upload an ssh key in your profile and get an shell prompt
with
: ssh pjotrpbl@shell.lugli.arvadosapi.com
: Linux ip-10-255-0-202 4.19.0-9-cloud-amd64 #1 SMP Debian 4.19.118-2+deb10u1 (2020-06-07) x86_64
It is a small Debian VM hosted on AWS somewhere. The PubSeq material
is mounted on ~/data/pubseq~. The log is in ~nohup.out~. Update/edit
the code (bh20-seq-resource git checkout) and restart the service (the
run script). The log says
: you should have permission to read the log (nohup.out) update / edit the code (bh20-seq-resource git checkout) and restart the service (the run script)
which means it will trigger the run on upload. The service is running as a
Python virtualenv:
: /data/pubseq/bh20-seq-resource/venv3/bin/python3 /data/pubseq/bh20-seq-resource/venv3/bin/bh20-seq-analyzer --no-start-analysis
and is restarted by a ~run~ script:
: /data/pubseq/run [options]
The run script kills the old process, sets up the API tokens, pulls
the git repo and starts a new run calling into
/data/pubseq/bh20-seq-resource/venv3/bin/bh20-seq-analyzer which is
essentially [[https://github.com/arvados/bh20-seq-resource/blob/2baa88b766ec540bd34b96599014dd16e393af39/bh20seqanalyzer/main.py#L354][monitoring]] for uploads.
On ~run --help~
#+begin_example
optional arguments:
-h, --help show this help message and exit
--uploader-project UPLOADER_PROJECT
--pangenome-analysis-project PANGENOME_ANALYSIS_PROJECT
--fastq-project FASTQ_PROJECT
--validated-project VALIDATED_PROJECT
--workflow-def-project WORKFLOW_DEF_PROJECT
--pangenome-workflow-uuid PANGENOME_WORKFLOW_UUID
--fastq-workflow-uuid FASTQ_WORKFLOW_UUID
--exclude-list EXCLUDE_LIST
--latest-result-collection LATEST_RESULT_COLLECTION
--kickoff
--no-start-analysis
--once
--print-status PRINT_STATUS
--revalidate
#+end_example
* Wiring up CWL
In above script ~bh20-seq-analyzer~ you can see that the [[https://www.commonwl.org/][Common
Workflow Language]] (CWL) gets [[https://github.com/arvados/bh20-seq-resource/blob/2baa88b766ec540bd34b96599014dd16e393af39/bh20seqanalyzer/main.py#L233][triggered]]; for example [[https://github.com/arvados/bh20-seq-resource/tree/master/workflows/fastq2fasta][fastq2fasta]] which
is part of the main repo. The actual script is in [[https://github.com/arvados/bh20-seq-resource/blob/master/workflows/fastq2fasta/fastq2fasta.cwl][fastq2fasta.cwl]] and
runs the following tools in sequence: bwa-mem, samtools-view,
samtools-sort, and bam2fasta.
It probably pays to familiarize yourself with CWL and its concepts. We
believe it has a lot going for it though CWL is some steps removed
from traditional shell scripts for running work flows. Main thing to
understand is that CWL is a separation of concerns, i.e.,
1. Data
2. Tools
3. Flow
and each of these are described separately. This contrasts largely
with shell scripts (though you can invoke shell scripts from CWL).
Also, CWL is written in JSON/YAML, which means everything can be parsed
as a tree and you can easily get visualisations such as
@@html:
@@
* The Arvados file interface
Arvados has the web server, but it also has a REST API and associated
command line tools. We are already using the [[https://github.com/arvados/bh20-seq-resource/blob/master/bh20sequploader/main.py#L27][API]] to upload data. If
you follow the pip or [[../INSTALL.md]] GNU Guix instructions for
installing Arvados API you'll find the following command line tools
(also documented [[https://doc.arvados.org/v2.0/sdk/cli/subcommands.html][here]]):
| Command | Description |
|---------+-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|
| arv-ls | list files in Arvados |
| arv-put | upload a file to Arvados |
| arv-get | get a textual representation of Arvados objects from the command line. The output can be limited to a subset of the object’s fields. This command can be used with only the knowledge of an object’s UUID |
Now, this is a public instance so we can use the tokens from
the [[https://github.com/arvados/bh20-seq-resource/blob/master/bh20sequploader/main.py#L16][uploader]].
#+BEGIN_SRC sh
export ARVADOS_API_HOST='lugli.arvadosapi.com'
export ARVADOS_API_TOKEN='2fbebpmbo3rw3x05ueu2i6nx70zhrsb1p22ycu3ry34m4x4462'
arv-ls lugli-4zz18-z513nlpqm03hpca
#+END_SRC
will list all files (the UUID we got from the Arvados results page). To
get the UUID of the files
#+BEGIN_SRC sh
curl https://lugli.arvadosapi.com/arvados/v1/config | jq .Users.AnonymousUserToken
env ARVADOS_API_TOKEN=5o42qdxpxp5cj15jqjf7vnxx5xduhm4ret703suuoa3ivfglfh \
arv-get lugli-4zz18-z513nlpqm03hpca
#+END_SRC
and fetch one listed JSON file ~chunk001_bin4000.schematic.json~ with
its listed UUID:
: arv-get 2be6af7b4741f2a5c5f8ff2bc6152d73+1955623+Ab9ad65d7fe958a053b3a57d545839de18290843a@5ed7f3c5
* The PubSeq Arvados shell
When you login to Arvados (you can request permission from us) it is
possible to upload an ssh key in your profile and get an shell prompt
with
: ssh pjotrpbl@shell.lugli.arvadosapi.com
: Linux ip-10-255-0-202 4.19.0-9-cloud-amd64 #1 SMP Debian 4.19.118-2+deb10u1 (2020-06-07) x86_64
It is a small Debian VM hosted on AWS somewhere. The PubSeq material
is mounted on ~/data/pubseq~. The log is in ~nohup.out~. Update/edit
the code (bh20-seq-resource git checkout) and restart the service (the
run script). The log says
: you should have permission to read the log (nohup.out) update / edit the code (bh20-seq-resource git checkout) and restart the service (the run script)
which means it will trigger the run on upload. The service is running as a
Python virtualenv:
: /data/pubseq/bh20-seq-resource/venv3/bin/python3 /data/pubseq/bh20-seq-resource/venv3/bin/bh20-seq-analyzer --no-start-analysis
and is restarted by a ~run~ script:
: /data/pubseq/run [options]
The run script kills the old process, sets up the API tokens, pulls
the git repo and starts a new run calling into
/data/pubseq/bh20-seq-resource/venv3/bin/bh20-seq-analyzer which is
essentially [[https://github.com/arvados/bh20-seq-resource/blob/2baa88b766ec540bd34b96599014dd16e393af39/bh20seqanalyzer/main.py#L354][monitoring]] for uploads.
On ~run --help~
#+begin_example
optional arguments:
-h, --help show this help message and exit
--uploader-project UPLOADER_PROJECT
--pangenome-analysis-project PANGENOME_ANALYSIS_PROJECT
--fastq-project FASTQ_PROJECT
--validated-project VALIDATED_PROJECT
--workflow-def-project WORKFLOW_DEF_PROJECT
--pangenome-workflow-uuid PANGENOME_WORKFLOW_UUID
--fastq-workflow-uuid FASTQ_WORKFLOW_UUID
--exclude-list EXCLUDE_LIST
--latest-result-collection LATEST_RESULT_COLLECTION
--kickoff
--no-start-analysis
--once
--print-status PRINT_STATUS
--revalidate
#+end_example
* Wiring up CWL
In above script ~bh20-seq-analyzer~ you can see that the [[https://www.commonwl.org/][Common
Workflow Language]] (CWL) gets [[https://github.com/arvados/bh20-seq-resource/blob/2baa88b766ec540bd34b96599014dd16e393af39/bh20seqanalyzer/main.py#L233][triggered]]; for example [[https://github.com/arvados/bh20-seq-resource/tree/master/workflows/fastq2fasta][fastq2fasta]] which
is part of the main repo. The actual script is in [[https://github.com/arvados/bh20-seq-resource/blob/master/workflows/fastq2fasta/fastq2fasta.cwl][fastq2fasta.cwl]] and
runs the following tools in sequence: bwa-mem, samtools-view,
samtools-sort, and bam2fasta.
It probably pays to familiarize yourself with CWL and its concepts. We
believe it has a lot going for it though CWL is some steps removed
from traditional shell scripts for running work flows. Main thing to
understand is that CWL is a separation of concerns, i.e.,
1. Data
2. Tools
3. Flow
and each of these are described separately. This contrasts largely
with shell scripts (though you can invoke shell scripts from CWL).
Also, CWL is written in JSON/YAML, which means everything can be parsed
as a tree and you can easily get visualisations such as
@@html: